| |
Correctly identifying various spectral lines and other features of the spectrum is the first step of performing accurate EDS analysis. However, in practice, it is easy to misidentify the X-ray lines based on preconceptions of the result and due to line overlaps even though the analyst think it should not. Line (or called peak) overlap makes identification difficult. Note that in some cases from EDS spectra alone, unambiguous peak assignment may be impossible. Especially, one of the most common errors made by EDS novices is misidentification of X-ray peaks.
For correct peak identification, one needs to follow the rules below:
i) The peak positions of EDS spectrometers should be calibrated accurately (see page1741). A miscalibrated system will shift X-ray peaks to improper energies, increasing the risk of misidentification. This is normally done by EDS manufacturers or by EM managers periodically.
ii) Record adequate counts statistically.
iii) Use automatic peak identification solution for the major peaks as a starting point. However, this auto peak identification should be considered only as a suggestion, and the analyst
should confirm the peak assignment as discussed here.
iv) The peaks of the X-ray families will appear in fixed ratios (see page4672 and page4637). Therefore, the possibility of peak misidentification will be lowered significantly if a full database (containing all family members) with all the X-ray peaks of the families from all elements is used.
v) Any weakly excited peaks will not be identified for a specific element unless its stronger peaks are visible.
vi) Elemental identification should begin with the major (primary) peak in the spectrum. All the lines for this specific element, e.g. escape and sum peaks, should then be located if they exist. Identification should then be performed to the next most intense peak, and so on. For instance, for the identification of primary X-ray Kα line, you need to confirm the presence of the corresponding Kβ and low-energy L
peak(s). You can download such database at excel file when you work on peak identification each time.
vii) Only the peaks that are statistically obvious should be considered for identification of characteristic X-rays. In general, the minimum amplitude of the peak P after background subtraction should be at least three times the standard deviation of the background counts NB in the
peak window (typically selected as 1.5 FWHM), i.e., P ≥ 3 (NB)1/2.
viii) For the identification of the elements that have low-energy X-rays only, a single peak will eventually be available. In this case, the analyst must taken advantage of every feature available, such as the asymmetry of the L and M peaks due to the relative heights and separations of the Lα–Lβ and Mα–Mβ peak pairs.
ix) Eliminate spurious X-rays, e.g. Cu, Fe, Ni, or Cr X-rays from SEM/TEM specimen chamber, column and apertures.
x) After the identifications above have been considered and performed, a few small features probably are still unidentified and may be coherent bremsstrahlung X-rays . This can be confirmed by recording another spectrum from the same area of the sample, but with either beam tilt or sample tilt. In either case, the position and probably the intensity of the peaks will change if they really belong to coherent bremsstrahlung.
xi) In order to correctly identify trace elements, one needs to follow:
xi.a) Record spectra that are statistically sufficient for trace peak identification.
xi.b) Systematically interpret the peaks from major elements and then remove all spectral features associated with these
major elements before attempting the possible trace elements. This process can be done by multiple linear least squares (MLLS) fitting technique.
xi.c)
Interpret the additional peaks associated with trace elements.
xii) An efficient way to identify if a peak belongs to a specific element (A) or not is that the spectrum is compared with a spectrum of element A by overlapping both spectra. The spectrum of element A can be taken from a known location of the same specimen or a different known specimen.
Almost all commercial EDS software usually includes two peak-identification methods:
i) Automatic Peak Identification.
ii) Manually Identifying Peaks. In this case, the rules listed above should be followed manually.
In the method of automated peak identification, the identification procedures in EDS measurements mainly consist of two parts:
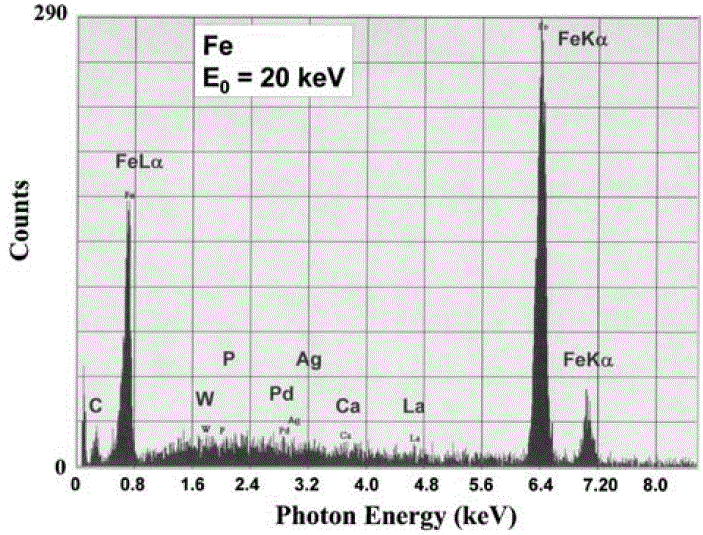
i) Peak searching algorithm locates peak channels for peaks above the minimum peak/noise threshold, P ≥ 3 (NB)1/2. This process is sensitive to the noise in the spectral background because random groupings of background counts can mimic a characteristic X-ray peak when the threshold is set too low. Figure 1747 was obtained when a short data acquisition time was used. The four peaks (Fe Kα, Fe Kβ, Fe Lα and C K) were really generated from the analyzing material; however, the other peaks (W, P, Pd, Ag, Ca and La) were false, which were misidentified from the noise in the background. Different artificial trace elements will show up because the noise in the background changes, when the data acquisition time increases. When the acquisition time is long enough so that P ≥ 3 (NB)1/2, the artificial peaks and thus the peak misidentification will then be eliminated. However, on many software, users are able to input their statistical threshold to define what constitutes a significant
peak above the random noise in the background.
| Figure 1747. Automated peak identification performed on a noisy EDS spectrum, taken at an accelerating voltage of 20 keV. [1] |
ii) Compare the found peaks with experimental database or theoretical calculations and closest match within ±ΔE (e.g., ±20 eV).
With the operation of automated peak identification, one needs to be aware:
i) The EDS system will produce erroneous identifications if it is slightly miscalibrated.
ii) It is a good idea to perform auto peak identification on specimens, whose compositions are known and are similar to that of the analyzing specimen, to confirm if the produced results are really correct.
iii) Auto ID software is not able to differentiate the overlap peaks with energy separations less than the energy resolution of the EDS system.
iv) Pile-up (sum) peaks are frequently misidentified by automatic identification
software.
v) The peak misidentifications are normally systematic and independent of the number of counts collected.
vi) Automation of peak identification is extremely vulnerable to misidentifying even major peaks, especially for trace elements. This happens with software from any EDS companies. In general, automated methods statistically fail to identify major peaks 3-5 %; however, some software fails more (up to 10%) and some does less. Furthermore, peak misidentification occurs more frequently when a low energy beam is used (e.g. for an accelerating voltage of ≤5 keV). Table 1747 lists some examples of common peak misidentifications from automatic method, and the full list for all the elements in periodic table can be found at excel file.
Table 1747. Common peak misidentifications from automatic method.
Energy range (keV) |
Peak energy (keV) |
Misidentified lines |
| < 1 |
0.390–0.395 |
N K (0.392); Sc Lα (0.395) |
| 0.510–0.525 |
O K (0.523); V Lα (0.511) |
| 0.670–0.710 |
F K (0.677); Mn Lα (0.636); Fe Lα (0.705) |
| 0.845–0.855 |
Zn LI; Ne Kα (0.848); Ni Lα (0.851) |
| 0.900–0.950 |
Cu Lα (0.928); Pr Mα (0.929) |
| 1 – 2 |
1.00–1.05 |
Na Kα (1.041); Zn Lα (1.012); Pm Mα (1.032) |
| 1.20–1.30 |
Br LI; Mg Kα (1.253); As Lα (1.282); Tb Mα (1.246) |
| 1.45–1.55 |
Br Ll; Al Kα (1.487); Br Lα (1.480); Yb Mα (1.521) |
| 1.69–1.80 |
Au Mζ; Si Kα (1.740); Rb Lα (1.694); Sr Lα (1.806); Tα Ma (1.709); W Mα (1.774) |
| 2 – 3 |
2.00–2.05 |
P Ka (2.013); Zr La (2.042)Pt Ma (2.048) |
| 2.10–2.20 |
Nb Lα (2.166); Au Mα (2.120); Hg Mα (2.191) |
| 2.28–2.35 |
S Kα (2.307); Mo Ll; Mo Lα (2.293); Pb Mα 2.342) |
| 2.40–2.45 |
Tc Lα (2.424); Bi Mα (2.419) |
| 2.60–2.70 |
Cl Kα (2.621); Rh Lα (2.696) |
| 2.95–3.00 |
Ar Kα (2.956); Ag Lα (2.983); Th Mα (2.996) |
| 3 – 4 |
3.10–3.20 |
Cd Lα (3.132); U Mα1 (3.170) |
| 3.25–3.35 |
K Kα (3.312); In Lα (3.285); U Mβ (3.336) |
| 3.60–3.76 |
K Kβ (3.589); Ca Kα (3.691); Sb Lα (3.605); Te Lα (3.769) |
| 4 – 5 |
4.05–4.15 |
Sc Kα (4.090); Xe Lα (4.111) |
| 4.45–4.55 |
Ti Kα (4.510); Ba Lα (4.467) |
| 4.84–4.95 |
Ti Kβ (4.931); V Kα (4.952); Ce Lα (4.840); Pr Lα (5.034) |
vii) Peak misidentification of minor and trace elements. The possibilities of peak misidentification of minor and trace family members can be even much higher than 10 % due to their lower concentrations. Because it will easily make extensive mistakes when EDS system automatically identifies trace constituent peaks, the default thresholds of EDS systems normally are not set to low enough to detect such low contents. For such cases, the analysts normally need to create their own appropriate analytical strategy manually.
viii) Peak misidentification of major elements. In this case, the escape peaks and sum peaks of the major elements are the main sources that cause misidentification unless the auto-identification procedure includes the energies of the relevant escape peaks and sum peaks in its database for each element. That is, their relatively low intensities are likely
to be misidentified as minor or trace element peaks. Furthermore, some software fails on given elements no matter how many counts are accumulated in the spectrum.
With automatic elemental identification of NSS software, Ce cannot be identified. Instead, the software incorrectly suggests Ti, Fe, Co, Os, and Pb everywhere in elemental maps.
[1] Dale E. Newbury, Mistakes Encountered During Automatic Peak Identification of Minor and Trace Constituents in Electron-Excited Energy Dispersive X-Ray Microanalysis, Scanning, 31, 1–11 (2009).
|
|